srm-eval
Project
High throughput sequencing (HTS) data volumes are rapidly growing and the need for their fast and accurate analysis becomes increasingly important. To accommodate this demand, a wide range of read alignment algorithms has been developed over the past years. Inside this spectrum those algorithms capable of producing spliced alignments form a particularly interesting subgroup of methods for dealing with RNA sequencing data, since their results are very valuable for downstream transcriptome analyses. The different approaches do not only differ significantly in running time, but also in result quality, robustness, and sensitivity to biased input data. Although many algorithms are in use, none of the current publications was accompanied by a comprehensive comparison of alignment performance and result quality.
Methods
Following the successful format of the EGASP workshop in 2005 (Guigo et al., Genome Biology, 2006), the RNASeq Genome Annotation Assessment Project (RGASP) was launched to assess the current progress of automatic gene building using RNA-Seq as its primary dataset. The goals of this community effort are to assess the success of computational methods to correctly map RNA-Seq data onto the genome, assemble transcripts and quantify their abundance in particular datasets. The input data originated from different sequencing platforms (Illumina, SOLiD, and Helicos) from H. sapiens, D. melanogaster and C. elegans. For these three organisms, which are also analyzed as part of the (mod)ENCODE project, high quality genome annotation is available which served as the references for the analysis.
As part of this effort, participants also submitted their alignments using a diversity of different methods, including BLAT, TopHat, PALMapper, and GSNAP. First, we compared the submitted alignments among each other. Figure 1 shows a pair-wise comparison of sanitized alignments for reads aligned to human chromosome 1, with respect to the agreement of the complete read alignment and with respect to the agreement of intron predictions inferred from spliced alignments. Moreover, we compared the submitted alignments with respect to different descriptive statistical criteria as, for instance, sensitivity and precision of intron recognition, mismatch and indel distribution, and the overlap to annotated transcript and exon boundaries. In Figure 2 we show the sensitivity and specificity of intron predictions inferred from spliced alignments for human (chromosome 1) where we used the ENCODE genome annotation as ground truth. For a selection of algorithms we carried out further analyses, based on the re-alignment of artificially generated reads. This approach has two advantages: the availability of the theoretically optimal alignment and the controllability of the error distribution in the generated data.
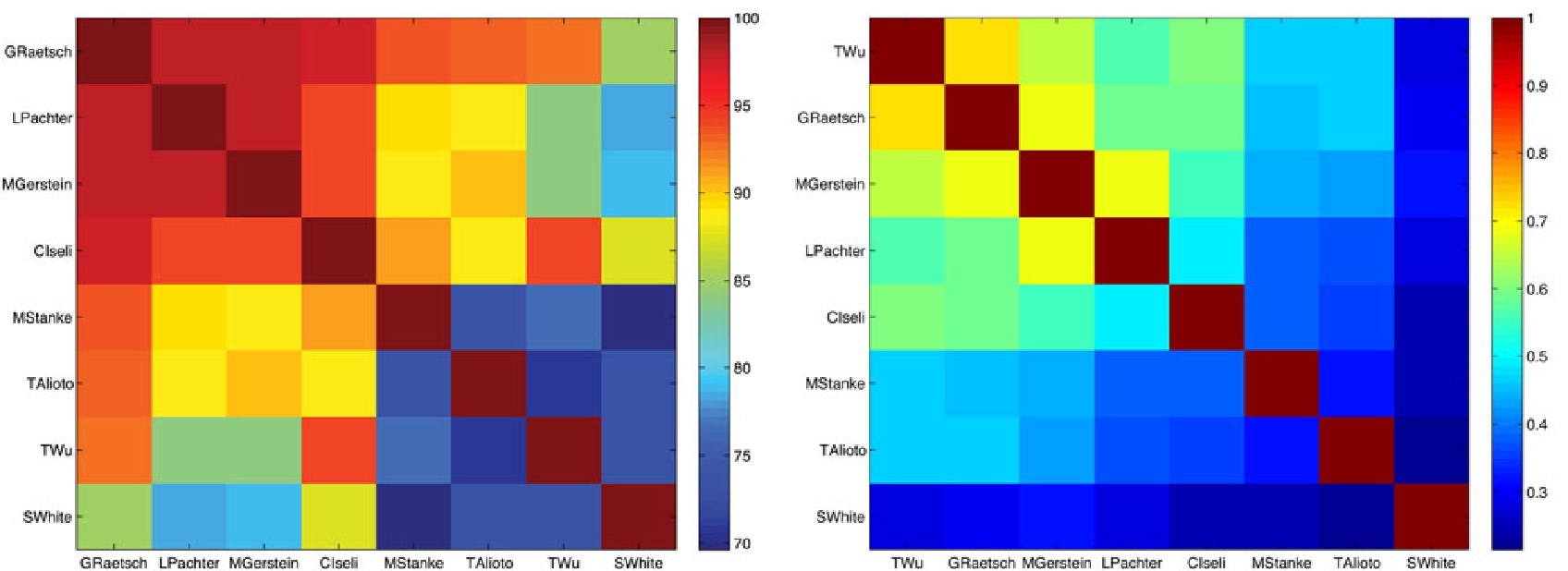
Figure 1. Pairwise comparison of submitted alignments: Agreement of the whole alignment for all reads with alignments in both submissions (left) and of intron predictions inferred from spliced alignments (right). Submissions are sorted by the degree of their agreement.
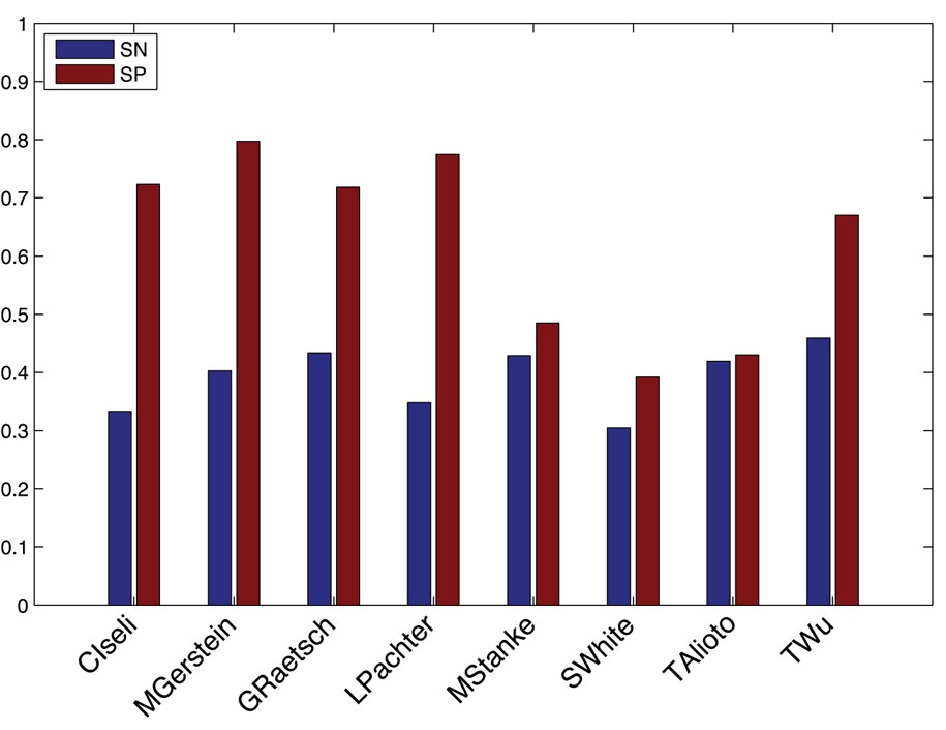
Figure 2: Sensitivity (SN) and specificity (SP) of intron predictions inferred from spliced alignments for human (chromosome 1), where the ENCODE annotation was used as ground truth.
Results
Our comparisons showed a great diversity in the behavior of the different alignment strategies. In particular, there is surprisingly small agreement between a subset of methods. We can show that the largest differences are the result of different alignment filtering strategies, which can, for instance, drastically increase the precision of intron predictions. The evaluations of the transcript annotations derived from these alignments performed within RGASP additionally allow us to correlate the alignment accuracy with the preciseness of exon, transcript, and gene prediction. We will discuss specific features of the different alignment strategies that most influence the success of subsequent analysis steps.
Software
The tremendous effects of filtering to alignment quality and results of downstream analyses have led to the development of a small software package: the Simple Alignment Filter Toolbox (SAFT). It is now available in a first release version. (Download SAFT-0.3)
The analysis tools developed during the project have also been incorporated into our publicly accessible and free to use Galaxy Webservice