
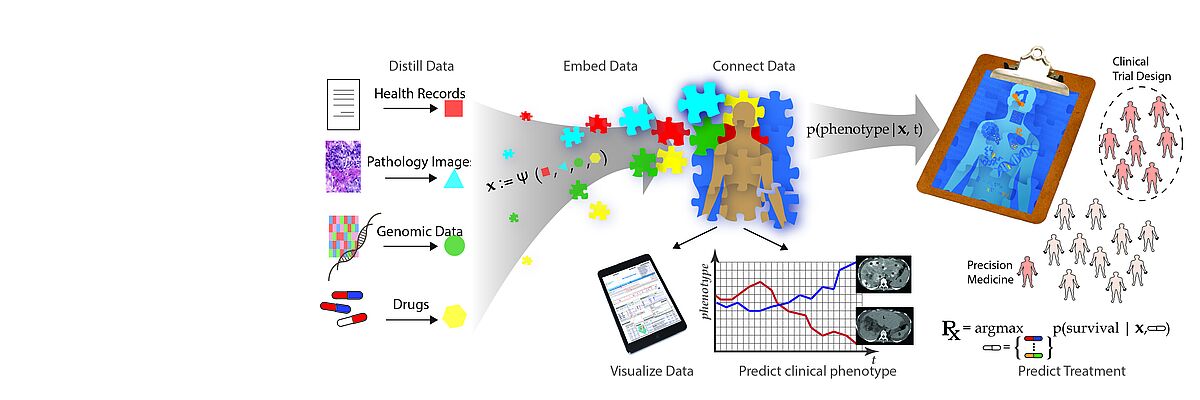
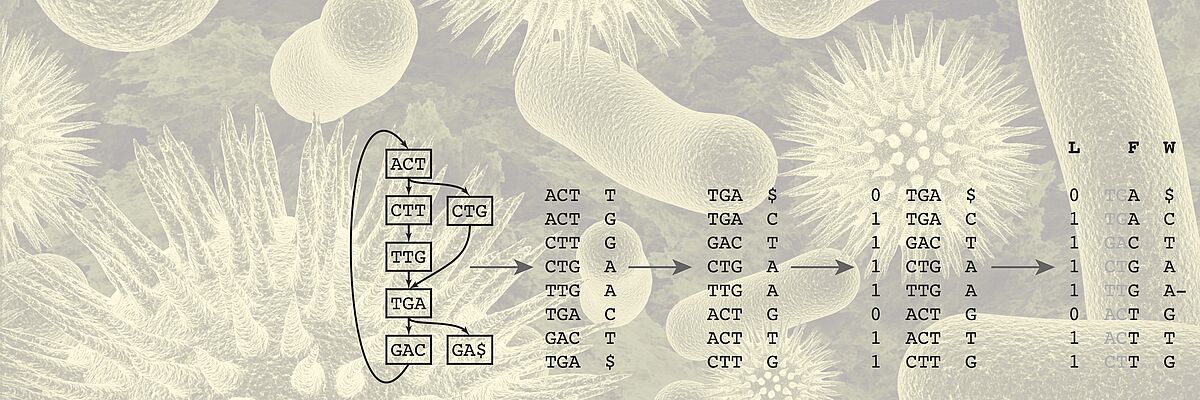

The BMI lab bridges research in Machine Learning and Sequence Analysis methodology research and its application to biomedical problems. We collaborate with biologists and clinicians to develop real-world solutions.
We work on research questions and foundational challenges in storing, analysing, and searching extensive heterogeneous and temporal data, especially in the biomedical domain. Our lab members address technical and non-technical research questions in collaboration with biologists and clinicians. At the research group’s core is an active knowledge exchange in both directions between the methods and the application-driven researchers.
The emergence of data-driven medicine leverages data and algorithms to shape how we diagnose and treat patients. Machine Learning approaches allow us to capitalise on the vast amount of data produced in clinical settings to generate novel biomedical insights and build more precise predictive models of disease outcomes and treatment efficacy.
We work towards this transformation mainly but not exclusively in two key areas. One key application area is the analysis of heterogeneous data of cancer patients. For Genomics, we develop algorithms for storing, compressing, and searching extensive genomics datasets. Another key area is the development of time series models of patient health states and early warning systems for intensive care units.
Publications
Abstract Spatial transcriptomics couples hematoxylin and eosin (H&E) tissue morphology with spatially resolved gene expression (GE). However, generative models that exploit this coupling to synthesize tissue images from transcriptomic profiles remain scarce. We present STMDiT (Spatial Transcriptomics and Morphology Diffusion Transformer), a diffusion transformer that synthesizes H&E histopathology patches conditioned jointly on morphological embeddings and transcriptomic profiles. Building on PixCell (Yellapragada et al., 2025), we integrate gene expression from a frozen CancerFoundation encoder (Theus et al., 2024) through adaptive layer normalization and per-block cross-attention, and we train under dual classifier-free guidance with independent modality dropout. On the 10x TuPro Visium melanoma cohort, GE conditioning improves both image quality over the no-GE PixCell-B baseline (best FID = 252.9 vs 330.7) and transcriptomic fidelity (best AUC = 0.267 vs 0.229, reaching 82% of the real-tile ceiling). Training with DeepSpot's predicted-transcriptomics pseudo-labels (PTPL) uniquely transfers zero-shot to TCGA SKCM, an out-of-distribution (OOD) H&E-only melanoma cohort: PTPL-XAttn-PMA-B reaches FID = 690.0, a 57-point improvement over the no-GE baseline (747.1), with a within-model GE-ablation effect of ΔOOD = +309.5, enabling virtual tissue synthesis beyond native spatial-transcriptomics coverage. Our results indicate that gene-expression conditioning produces morphologically distinct tissue images and supports virtual tissue simulation for hypothesis testing in computational pathology. Code availability: https://github.com/ratschlab/stmdit
Authors Pantelis Vlachas, Kalin Nonchev, Viktor H Koelzer, Gunnar Rätsch
Submitted ICML 2026 Multi-modal Foundation Models and Large Language Models for Life Sciences
Abstract The growing number of spatial transcriptomics (ST) datasets enables comprehensive multi-modal characterization of cell types across diverse biological and clinical contexts. However, integration across patient cohorts remains challenging, as local microenvironment, patient-specific variability, and technical batch effects can dominate signals. Here, we hypothesize that combining specialized transcriptomics correction methods with deep representation learning can jointly align morphology, transcriptomics, and spatial information across multiple tissue samples. This approach benefits from recent transcriptomics and pathology foundation models, projecting cells into a shared embedding space where they cluster by cell type rather than dataset-specific conditions. Applying this framework to 18 skin melanoma, 12 human brain, and 4 lung cancer datasets, we demonstrate that it outperforms conventional batch-correction approaches by 58%, 38%, and 2-fold, respectively. Together, this framework enables efficient integration of multi-modal ST data across modalities and samples, facilitating the systematic discovery of conserved cellular programs and spatial niches while remaining robust to cohort-specific batch effects. Code availability https://github.com/ratschlab/aestetik
Authors Justina Dai, Kalin Nonchev, Viktor H Koelzer, Gunnar Rätsch
Submitted ICLR 2026 LMRL
Abstract Clinical time series data are critical for patient monitoring and predictive modeling. These time series are typically multivariate and often comprise hundreds of heterogeneous features from different data sources. The grouping of features based on similarity and relevance to the prediction task has been shown to enhance the performance of deep learning architectures. However, defining these groups a priori using only semantic knowledge is challenging, even for domain experts. To address this, we propose a novel method that learns feature groups by clustering weights of feature-wise embedding layers. This approach seamlessly integrates into standard supervised training and discovers the groups that directly improve downstream performance on clinically relevant tasks. We demonstrate that our method outperforms static clustering approaches on synthetic data and achieves performance comparable to expert-defined groups on real-world medical data. Moreover, the learned feature groups are clinically interpretable, enabling data-driven discovery of task-relevant relationships between variables.
Authors Fedor Sergeev, Manuel Burger, Polina Leshetkina, Vincent Fortuin, Gunnar Rätsch, Rita Kuznetsova
Submitted ML4H 2025 (PMLR)
Abstract CRISPR-based genetic perturbation screens paired with single-cell transcriptomic readouts (Perturb-seq) offer a powerful tool for interrogating biological systems. Yet the resulting datasets are heterogeneous—particularly in vivo—and currently used cell-level perturbation labels reflect only CRISPR guide RNA exposure rather than perturbation state; further, many perturbations have a minimal effect on gene expression. For perturbations that do alter the transcriptomic state of cells, intracellular guide RNA abundance exhibits a dose-response association with perturbation efficacy. We combine (i) per-perturbation, expression-only classifiers trained with non-negative negative–unlabeled (nnNU) risk to yield calibrated scores reflecting the perturbation state of single cells and (ii) a monotone guide abundance prior to yield a per-cell pseudo-posterior that supports both assignment of perturbation probability and selection of affected gene features. To obtain a low-dimensional representation that allows for the accurate reconstruction of gene-level marginals for counterfactual decoding, we train an autoencoder with a quantile–hurdle reconstruction loss and feature-weighted emphasis on perturbation-affected genes. The result is a perturbation-aware latent embedding amenable to downstream trajectory modeling (e.g., optimal transport or flow matching) and a principled probability of perturbation for each non-control cell derived jointly from its guide counts and transcriptome.
Authors Florian Hugi, Tanmay Tanna, Randall J. Platt, Gunnar Rätsch
Submitted NeurIPS 2025 AI4D3
Abstract Spot-based spatial transcriptomics (ST) technologies like 10x Visium quantify genome-wide gene expression and preserve spatial tissue organization. However, their coarse spot-level resolution aggregates signals from multiple cells, preventing accurate single-cell analysis and detailed cellular characterization. Here, we present DeepSpot2Cell, a novel DeepSet neural network that leverages pretrained pathology foundation models and spatial multi-level context to effectively predict virtual single-cell gene expression from histopathological images using spot-level supervision. DeepSpot2Cell substantially improves gene expression correlations on a newly curated benchmark we specifically designed for single-cell ST deconvolution and prediction from H&E images. The benchmark includes 20 lung, 7 breast, and 2 pancreatic cancer samples, across which DeepSpot2Cell outperformed previous super-resolution methods, achieving respective improvements of 46%, 65%, and 38% in cell expression correlation for the top 100 genes. We hope that DeepSpot2Cell and this benchmark will stimulate further advancements in virtual single-cell ST, enabling more precise delineation of cell-type-specific expression patterns and facilitating enhanced downstream analyses. Code availability: https://github.com/ratschlab/DeepSpot
Authors Kalin Nonchev, Glib Manaiev, Viktor H Koelzer, Gunnar Rätsch
Submitted NeurIPS 2025 Imageomics


