PALMA: mRNA to Genome Alignments using Large Margin Algorithms
by Uta Schulze, Bettina Hepp, Cheng Soon Ong and Gunnar Rätsch.
The paper is published in the Bioinformatics journal (Bioinformatics 2007 23(15):1892-1900; doi:10.1093/bioinformatics/btm275). Download the preprint here. An earlier version of this work was published at GCB 2006.
Abstract
- Motivation:
- Despite many years of research on how to properly align sequences in the presence of sequencing errors, alternative splicing and micro-exons, the correct alignment of mRNA sequences to genomic DNA is still a challenging task.
- Results:
- We present a novel approach based on large margin learning that combines accurate splice site predictions with common sequence alignment techniques. By solving a convex optimization problem, our algorithm -- called PALMA -- tunes the parameters of the model such that true alignments score higher than other alignments. We study the accuracy of alignments of mRNAs containing artificially generated micro-exons to genomic DNA. In a carefully designed experiment, we show that our algorithm accurately identifies the intron boundaries as well as boundaries of the optimal local alignment. It outperforms all other methods: for 5702 artificially shortened EST sequences from C. elegans and human it correctly identifies the intron boundaries in all except two cases. The best other method is a recently proposed method called exalin which misaligns 37 of the sequences. Our method also demonstrates robustness to mutations, insertions and deletions, retaining accuracy even at high noise levels.
Software
PALMA aligns two genomic sequences in an optimal way according to its underlying algorithm and trained parameters. The main python script takes two FASTA files containing the target (e.g. a DNA sequence, part of the genome) and query sequences (e.g. a cDNA or EST sequence). It creates an alignment using dynamic programming (written in c++), and returns the alignment in a psl like format [Ken02]. The algorithms computes optimal local alignments, so if no alignment has been found it is because no alignment got a sufficiently high alignment score.
Licensing Information
PALMA is licensed under the GPL version 2 or any later version (cf. COPYING).
Releases
Download the source code of PALMA here (tested on Mac OS X and Linux):
- palma-0.3.7.tar.gz This version has parameters for several other organism (see below). The default version now only contains parameters for C.elegans. Several changes were done to track changes in the shogun interface. (19 October 2007, 4.4M)
- palma-0.3.6.tar.gz Extension to support blat-like format and bug fixes; 22 May 2007 (8.6MB)
- palma-0.3.4.tar.gz (This uses shogun for splice sites; 29 March 2007)
- palma-0.2.tar.gz (This is a version of Palma w/o SS.)
Organism specific parameters
Download the parameters for the organism you are studying, and add to the palma/data directory. E.g. use option --organism=drosophila to use the drosophila parameters. Please rename the downloaded file such that the file name is as below.:
Contact
In case of comments, problems, questions etc. feel free to contact Gunnar Rätsch.
Results
Comparison of different methods for aligning mRNAs to genomic DNA. 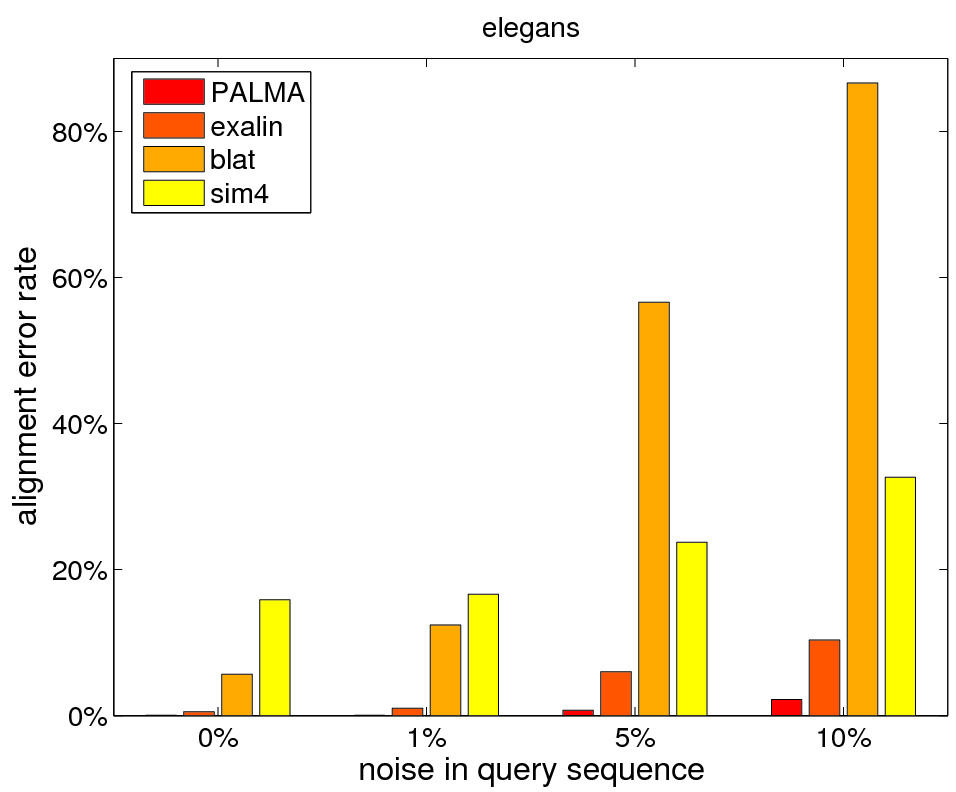
We considered the particularly difficult task of aligning exon triples with short middle exons (2-50nt) in the presence of noise. Almost all methods perform reasonably well when the query perfectly matches the target. For blat and sim4 the error rates drastically increase when adding noise to the query sequence. Only exalin and PALMA (with and without splice site information) have low error rates for noise levels of at most 20%. However, only PALMA (with splice sites) achieves 0% error rate for aligning queries with up to 10% noise. When deleting, inserting or mutating up to 50% of the query sequence, PALMA (with splice sites) still aligns 95% of all sequences correctly, while the other methods achieve less than 36% accuracy. For these large noise levels the splice site information helps to reduce the error rate considerably. But also in the low noise cases the splice site predictions help to accurately identify very short exons that can be found ambiguously in the intronic regions (0.4% of the test sequences).
Parameters used for the microexon experiment in the paper:
- elegans_micro_WS150_ss=0_il=0.param.bz2 (1k)
- elegans_micro_WS150_ss=0_il=1.param.bz2 (1k)
- elegans_micro_WS150_ss=1_il=0.param.bz2 (4.3M)
- elegans_micro_WS150_ss=1_il=1.param.bz2 (4.3M)
- human_micro_hg17_ss=1_il=1.param.bz2 (14.6M)
The datasets used for evaluation:
| training | elegans-training.tar.gz | human-training.tar.gz |
| validation | elegans-validation.tar.gz | human-validation.tar.gz |
| test | elegans-test.tar.gz | human-test.tar.gz |
Supplementary material
Training the splice site model and also the large margin combination requires separate sequence data sets. For the splice site model, we used genes that were EST confirmed but without full length cDNA support (set 1). We consider a random subset of 40% of all cDNA confirmed genes without evidence for alternative splicing for training the large margin combination (set 2). The remaining 20% and 40% were used for hyper-parameter tuning (set 3) and final evaluation (set 4) respectively.
For details see below.
Processing of Sequence Databases
We collected all known C. elegans ESTs from Wormbase [HCC04] (release WS120; 236,893 sequences) and dbEST [BT93] (as of February 22, 2004; 231,096 sequences). Using blat [Ken02] we aligned them against the genomic DNA (release WS120). The alignment was used to confirm exons and introns. We refined the alignment by correcting typical sequencing errors, for instance by removing minor insertions and deletions. If an intron did not exhibit the consensus GT/AG or GC/AG at the 5' and 3' ends, then we tried to achieve this by shifting the boundaries up to 2 base pairs (bp). If this still did not lead to the consensus, we split the sequence into two parts and considered each subsequence separately. In a next step we merged consistent alignments, if they shared at least one complete exon or intron. This lead to a set of 124,442 unique EST-based sequences.
We repeated the above procedure with all known cDNAs from Wormbase (release WS120; 4,855 sequences). These sequences only contain the coding part of the mRNA. We used their ends as annotation for start and stop codons.
We clustered the sequences in order to obtain independent training, validation and test sets. In the beginning each of the above EST and cDNA sequences were in a separate cluster. We iteratively joined clusters, if any two sequences from distinct clusters match to the same genomic location (this includes many forms of alternative splicing). We obtained 21,086 clusters, while 4072 clusters contained at least one cDNA.
For set 1 we chose all clusters not containing a cDNA (17215), for set 2 we chose 40% of the clusters containing at least one cDNA (1536). For set 3 we used 20% of clusters with cDNA (775). The remaining 40% of clusters with at least one cDNA (1,560) were used as set 4. Sets 2-4 were filtered to remove confirmed alternative splice forms. This left 1,177 cDNA sequences for testing in set 4 with an average of 4.8 exons per gene and 2,313bp from the 5' to the 3' end.
Artificial Microexon Dataset
Based on sets 2-4 described above we created sets of consecutive exon triples from the confirmed transcripts in these sets. This lead to 4604, 2257 and 4358 triples. In a first processing step we shortened the middle exons to a random length between 2nt and 50nt (uniformly distributed). To do so, we removed the correct number of nucleotides from the center of the middle exon - from the query as well as the DNA. This leaves the splice sites mostly functional. In a second step we added varying amounts of noise. For a given noise level p and a query sequence of length L, we first replaced p*L/3 positions with a random letter (Sigma={A,C,G,T,N}). Then we deleted the same number of non-overlapping positions in the sequence and added the same number of random nucleotides at random positions. We used p=0%, 1%, 10%, 20%, 50%.
Datasets from GCB 06 paper
These archives contain fasta-files including the DNA sequences and query sequences (EST/cDNA) for all noise levels. The true exon boundaries are provided. See the README files (example) for details.
- Training set: training.tar.gz (4.6 MB)
- Validation set: validation.tar.gz (2.3 MB)
- Test set: test.tar.gz (4.3 MB)
References
| [BT93] | M.S. Boguski and T.M. Lowe C.M. Tolstoshev. dbEST–Database for ”Expressed Sequence Tags”. Nat Genet., 4(4):332–3, 1993. |
| [HCC04] | T.W. Harris, N. Chen, F. Cunningham, et al. WormBase: a multi-species resource for nematode biology and genomics. Nucl. Acids Res., 32, 2004. Database issue:D411-7. |
| [Ken02] | (1, 2) W.J. Kent. BLAT–the BLAST-like alignment tool. Genome Res, 12(4):656–664, April 2002. |