NMD
Summary
The nonsense-mediated decay (NMD) surveillance pathway can recognize erroneous transcripts and physiological mRNAs, such as precursor mRNA alternative splicing (AS) variants. Currently, information on the global extent of coupled AS and NMD remains scarce and even absent for any plant species. To address this, we conducted transcriptome-wide splicing studies using Arabidopsis thaliana mutants in the NMD factor homologs UP FRAMESHIFT1 (UPF1) and UPF3 as well as wild-type samples treated with the translation inhibitor cycloheximide. Our analyses revealed that at least 17.4% of all multiple-exon, protein-coding genes produce splicing variants that are targeted by NMD. Moreover, we provide evidence that UPF1 and UPF3 act in a translation-independent mRNA decay pathway. Importantly, 92.3% of the NMD-responsive mRNAs exhibit classical NMD-eliciting features, supporting their authenticity as direct targets. Genes generating NMD-sensitive AS variants function in diverse biological processes, including signaling and protein modification, for which NaCl stress–modulated AS- NMD was found. Besides mRNAs, numerous noncoding RNAs and transcripts derived from intergenic regions were shown to be NMD responsive. In summary, we provide evidence for a major function of AS-coupled NMD in shaping the Arabidopsis transcriptome, having fundamental implications in gene regulation and quality control of transcript processing.
Supplementary Data
Computational Methods
The computational pipeline developed for NMD analysis is depicted below:
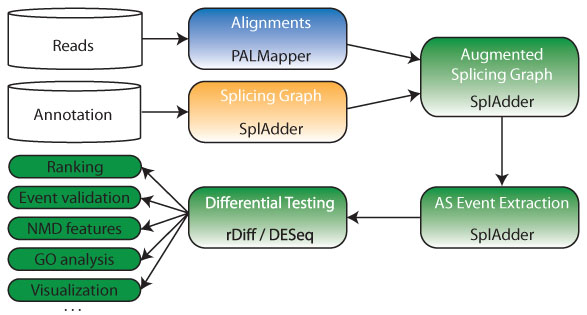
All tools as well as the pipeline as a whole are applicable to RNA-Seq data from any organism.
- A very sensitive RNA-Seq alignment has been performed using PALMapper, an aligner that can identify splice junctions not present in the annotated reference
- The number of uniquely mappable reads has been improved by multiple mapper resolution using the RNA-geeq toolbox
- Differential testing taking biological variants into account and making optimal use of the available replicates have been carried out with rDiff
- Testing for differential gene expression is based on DESeq
- Prediction of novel transcripts was done using MiTie
- All additional scripts in Matlab, Python, and bash are availanble from the authors upon request.