MMR
Mapping high throughput sequencing data to a reference genome is an essential step for most existing analysis pipelines aiming at the computational analysis of genome and transcriptome sequencing data. Breaking ties between equally likely mapping locations still poses a severe problem not only during the alignment phase but also has significant impact how the data can be used for further analyses. We present the multimapper resolution (MMR) tool that infers an optimal mapping location only on the distribution of mapped reads.
Principle
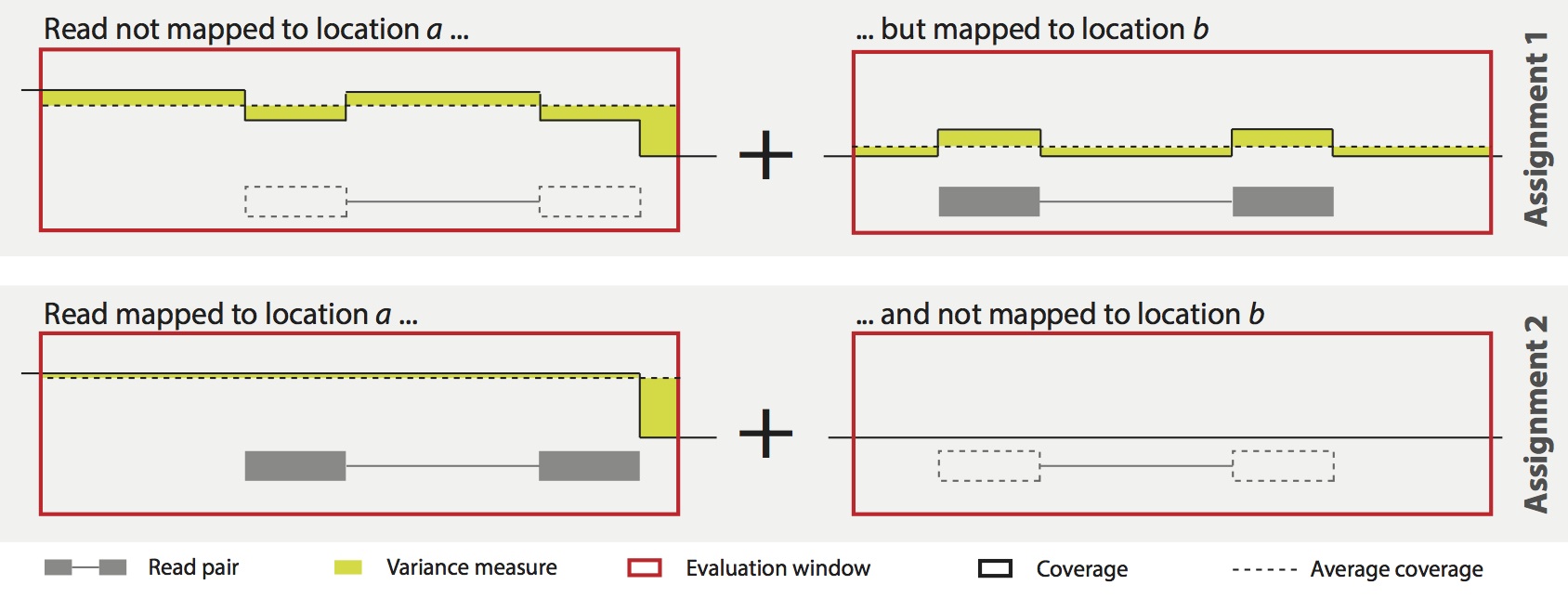
We follow a quite simple strategy that locally minimizes the variance of position wise coverage within windows around two possible mapping locations. Comparing the local "roughness" around two possible mappings, we can iteratively optimize all mappings to converge to a globally "smooth" coverage. Additional information, e.g., transcript structure, to accounting for expected differences in local coverage can be incorporated. The following figure illustrates the MMR principle.
Availability
The source code is public under GPLv3 and can be downloaded from GitHub.
Datasets
MMR has been evaluated on various sets of simulated data. Raw fastq files, alignments with PALMapper and TopHat2 as well as quantification results with rQuant and Cufflinks are available for download:
| Dataset | Link |
|---|---|
| Simulation data and results on A. thaliana WGS | Go to A thaliana data |
| Simulation data and results on human RNA-Seq (32nt) | Go to human 32nt data |
| Simulation data and results on human RNA-Seq (51nt) | Go to human 51nt data |
| Simulation data and results on human RNA-Seq (76nt) | Go to human 76nt data |
| Simulation data and results on human RNA-Seq (101nt) | Go to human 101nt data |
| Simulation data and results on human RNA-Seq (151nt) | Go to human 151nt data |