mGene: A Novel Discriminative Gene Finding System
News
| July 9, 2010: | Talk on "Next Generation Genome Annotation with mGene.ngs" at ISCB Student Council Symposium |
|---|---|
| June 23, 2009: | Paper describing mGene accepted for publication in Genome Research [3] |
| June 3, 2009: | Paper describing mGene.web published in Nucleic Acids Research [1]. |
| June 1, 2009: | Software release mgene-0.1.0-beta is available for download at http://mgene.org/download |
| March 1, 2009: | mGene.web goes live, see http://mgene.org/web |
Web interface: mGene.web
- Genome-wide prediction of protein-coding genes for eukaryotes (currently limited to genome sizes of several hundred Mbp). For this task we provide several pre-trained models (see below for the current list of organisms).
- Training the gene finder on your own data to generate a predictive model for newly sequenced genomes
- Evaluation / comparison of gene annotations
- Training and prediction of individual sensors, including splice sites, transcription start sites, etc.
Download mGene
Prediction Results
mGene at the nGASP competition
Recently we have participated in the nGASP competition launched by the Wormbase Consortium [2]. Compared to state-of-the-art gene finding systems mGene achieved excellent performance values. The results for the ab-initio category are shown below.
| Nucleotide | Exon | Transcript | Gene | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | Sn | Sp | Avg | Sn | Sp | Avg | Sn | Sp | Avg | Sn | Sp | Avg |
| mGene.init | 96.8 | 90.9 | 93.8 | 85.1 | 80.2 | 82.6 | 49.6 | 42.3 | 45.9 | 60.7 | 42.3 | 51.5 |
| mGene.init (dev) | 96.9 | 91.6 | 94.2 | 84.2 | 78.6 | 81.4 | 44.3 | 38.7 | 41.5 | 54.3 | 40.1 | 47.2 |
| Craig | 95.5 | 90.9 | 93.2 | 80.3 | 78.2 | 79.2 | 35.7 | 35.4 | 35.6 | 43.7 | 35.4 | 39.6 |
| Fgenesh | 98.2 | 87.1 | 92.7 | 86.4 | 73.6 | 80.0 | 47.1 | 34.1 | 40.6 | 57.7 | 34.1 | 45.9 |
| Augustus | 97.0 | 89.0 | 93.0 | 86.1 | 72.6 | 79.3 | 52.9 | 28.6 | 40.8 | 64.4 | 34.5 | 49.4 |
Table 1: Shown are sensitivity (Sn), specificity (Sp) and their average (Avg), each in percent, on nucleotide, exon, transcript and gene levels. The predictions of mGene.init were prepared after the deadline but strictly adhering to the rules and conditions of the nGASP challenge. The evaluation is based on the submitted sets of the participants and performed with our own routine. The numbers slightly deviate from the official nGASP evaluation on the transcript and gene level due to minor differences in the evaluation criteria. These differences, however, do not change the ranking.
More details on the mGene versions applied in nGASP, which also include a conservation based version, as well as a version that uses EST alignments, see mgene at nGASP competition
Genome-wide mGene predictions on five nematodes
We have used the mGene model trained on C. elegans for the genome (re-)annotation of five nematode genomes including: C. elegans, C. brenneri, C. japonica, C. remanei, and C. briggsae. In an assessment of the quality of several available annotations for these genomes, we find that mGene's predictions are most accurate. The gene predictions have been included in the wormbase annotation available at http://www.wormbase.org and http://mgene.org/predictions (also http://ftp.raetschlab.org/resources/mgene/genome_research_2009). They allow us to compare the resulting proteomes among these organisms and to the known protein universe, thereby identifying many species/lineage-specific gene inventions [3].
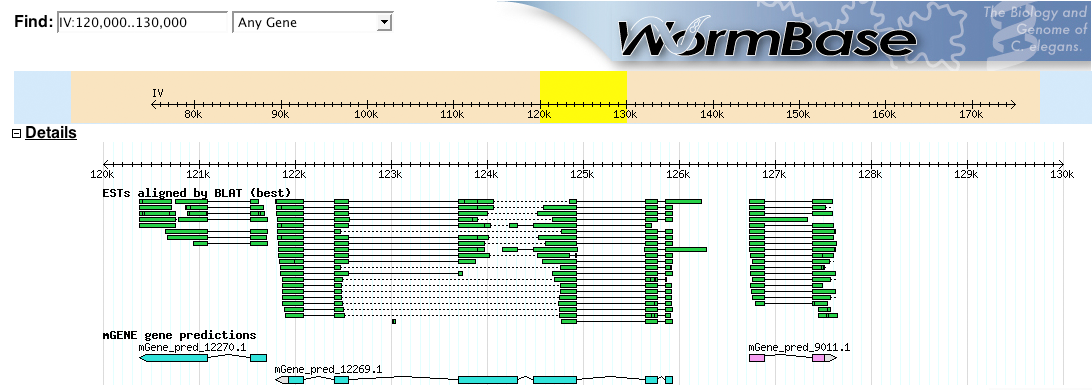
Figure 1: Wormbase screenshot, showing aligned ESTs and three genes predicted by mGene.
Training mGene on other organisms
We are currently training mGene on several other organisms. Here is a list of organisms, for which we can provide initial models:
Preliminary prediction performance measurements can be found here
Methods
We tackle the gene prediction problem taking a two-layered approach. In a first step, state-of-the-art kernel machines are employed to detect signal sequences in genomic DNA (like splice sites or transcription start sites) and to discriminate the content of different DNA sequences (like coding exons, introns, etc.). In a second step their outputs are combined to predict whole gene structures. In this step we use a discriminative training approach based on Hidden semi-Markov support vector machines [3] [4] [5]. A more detailed description is also provided here.
Contact
In case of comments, problems, questions etc. feel free to contact
References
| [1] | (1, 2) Schweikert, G, Behr, J, Zien, A, Zeller, G, Ong, CS, Sonnenburg, S, and Rätsch, G (2009). mGene.web: a web service for accurate computational gene finding. Nucleic Acids Research, Web Server Issue. |
| [2] | Coghlan et al. nGASP: the nematode genome annotation assessment project. BMC Bioinformatics, 2008. |
| [3] | (1, 2, 3) Schweikert, G, Zien, A, Zeller, G, Behr, J, Dieterich, C, Ong, CS, Philips, P, De Bona, F, Hartmann, L, Bohlen, A, Krüger, N, Sonnenburg, S, and Rätsch, G. mGene: Accurate SVM-based Gene Finding with an Application to Nematode Genomes. Genome Research, Advance Access June 29, 2009. |
| [4] | Raetsch and Sonnenburg. Large Scale Semi Hidden Markov SVMs. In Advances in Neural Information Processing Systems 19, Cambridge, MA, 2006. MIT Press. |
| [5] | Raetsch et al. Improving the Caenorhabditis elegans Genome Annotation Using Machine Learning, PLoS Computational Biology, 3(2):e20, 2007 |