For a nice and refreshing read in the summer heat, ETH News has kindly summarized it here or dare to go full length.
We will also present the work at this years ASHG 2018 in San Diego on Saturday, October 20, 11:40 AM–1:00 PM in the "108. Featured Plenary Abstract Session III".
Highlights
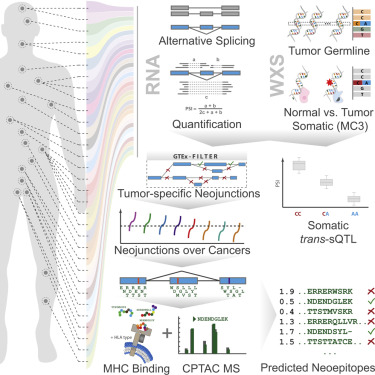
- Systematic analysis of alternative splicing landscape across 8,705 cancer patients
- Somatic trans-sQTL analysis identifies drivers of global splicing aberrations
- Many tumors contain numerous neojunctions not typically found in normal samples
- Neojunctions can be confirmed by MS and form a class of potential neoantigens
Summary Our comprehensive analysis of alternative splicing across 32 The Cancer Genome Atlas cancer types from 8,705 patients detects alternative splicing events and tumor variants by reanalyzing RNA and whole-exome sequencing data. Tumors have up to 30% more alternative splicing events than normal samples. Association analysis of somatic variants with alternative splicing events confirmed known trans associations with variants in SF3B1 and U2AF1 and identified additional trans-acting variants (e.g., TADA1, PPP2R1A). Many tumors have thousands of alternative splicing events not detectable in normal samples; on average, we identified ≈930 exon-exon junctions (“neojunctions”) in tumors not typically found in GTEx normals. From Clinical Proteomic Tumor Analysis Consortium data available for breast and ovarian tumor samples, we confirmed ≈1.7 neojunction- and ≈0.6 single nucleotide variant-derived peptides per tumor sample that are also predicted major histocompatibility complex-I binders (“putative neoantigens”).